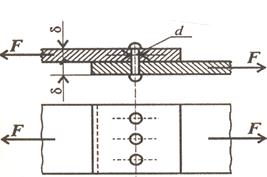
Силы межмолекулярного взаимодействияВ 1873 г. голландский ученый И.Ван-дер-Ваальс предположил, что существуют силы, обусловливающие притяжение между молекулами. Эти силы позднее получили название вандерваальсовых сил. Они включают в себя три составляющие: диполь-дипольное, индукционное и дисперсионное взаимодействия. Характерными чертами этих сил являются аддитивность (действие их складывается) и ненасыщаемость (число молекул, ориентирующихся около диполей, неопределенно). Диполь-дипольное или ориентационное взаимодействие. При сближении полярных молекул они ориентируются таким образом, чтобы положительная сторона одного диполя была ориентирована к отрицательной стороне другого диполя. Возникающие в результате этого ориентационные силы (силы Кеезона) будут стремиться удерживать молекулы вместе. Индукционное взаимодействие. Диполи могут воздействовать на неполярные молекулы, превращая их в индуцированные (наведенные диполи). Между постоянными и наведенными диполями возникает притяжение. Энергия индукционного взаимодействия возрастает с увеличением поляризуемости молекул, т.е. способности молекулы к образованию диполя под воздействием электрического поля. Поляризуемость в однотипных молекулах растет с увеличением их размеров. Энергия индукционного взаимодействия значительно меньше энергии ориентационного. Дисперсионное взаимодействие. Дисперсионные силы возникают потому, что в атоме существуют колебательные движения ядра и электронов относительно друг друга. В результате в атоме возникают как бы флуктуирующие (виртуальные) диполи, которые в свою очередь индуцируют мгновенные диполи у соседних молекул. Движение мгновенных диполей становится согласованным, их появление и распад происходит синхронно. В результате взаимодействия мгновенных диполей энергия системы понижается. Энергия дисперсионного взаимодействия пропорциональна поляризуемости молекул и обратно пропорциональна расстоянию между центрами частиц. Для неполярных молекул дисперсионное взаимодействие является единственной составляющей вандерваальсовых сил. Энергия дисперсионного взаимодействия меньше энергии ориентационного и индукционного взаимодействия. Для дисперсионных сил характерно быстрое убывание с расстоянием (энергия взаимодействия обратно пропорциональна шестой степени расстояния между молекулами), поэтому дисперсионное взаимодействие проявляется только при тесном сближении частиц. Этот вид межмолекулярного взаимодействия универсален, т.е. он возникает между любыми частицами, независимо от наличия у них заряда или диполей. Например, только дисперсионным взаимодействием можно объяснить притяжение между атомами (одноатомными молекулами) инертных газов в жидкой и твердой фазах; если бы не дисперсионное взаимодействие, то инертные газы при любой температуре оставались бы газообразными. Силы межмолекулярного взаимодействия примерно на два порядка слабее сил, удерживающих атомы при возникновении между ними обычной химической связи: их энергия составляет всего несколько кДж/моль, обычных же химических связей - сотни кДж/моль. Поэтому для преодоления сил межмолекулярного взаимодействия требуется гораздо меньше энергии, чем для разрыва ковалентной или ионной связи. 15)
Первое начало термодинамики. Первое начало термодинамики по существу выражает закон сохранения энергии. Для системы, окруженной замкнутой границей, через которую не происходит переноса вещества, справедливо соотношение
где U 1 и U 2 – энергии системы в состояниях 1 и 2; Q – теплота, полученная от внешних источников; W – работа, совершенная системой над внешними телами в процессе, посредством которого система переходит из состояния 1 в состояние 2. Если процесс – химическая реакция, то обычно ее проводят в таких условиях, чтобы можно было отделить энергию химического превращения от энергии, связанной с одновременными изменениями температуры или давления. Поэтому энергию (теплоту) химической реакции обычно определяют в условиях, в которых продукты находятся при тех же температуре и давлении, что и реагенты. Энергия химической реакции тогда определяется теплотой Q, полученной от окружающей cреды или переданной ей. Измерение Q может быть проведено с помощью калориметра подходящего типа. Реакцию можно было бы провести, например, в металлическом сосуде, погруженном в теплоизолированный объем воды, изменение температуры которой (обычно на несколько градусов) соответствует теплоте реакции. Для количественных измерений калориметр обычно градуируют с помощью независимого электронагревателя или проведения в сосуде химической реакции, теплота которой известна. Медленные реакции особенно трудны для калориметрических измерений, поскольку нужны сложные меры предосторожности для защиты калориметра от теплообмена с окружающей средой. Так называемый адиабатический калориметр целиком погружается в изотермическую оболочку с независимым нагревом, температура которой во время опыта поддерживается как можно более близкой к температуре внутри калориметра. Реакции, высвобождающие теплоту (отрицательная Q в уравнении (1)), называются экзотермическими, а реакции, в процессе которых теплота поглощается, – эндотермическими. Как показывает уравнение (1), внутренняя энергия реагирующей системы определяется не только количеством высвобожденной или поглощенной теплоты. Она также зависит от того, сколько энергии система затрачивает или приобретает посредством произведенной работы. При постоянном давлении p полная работа, совершенная системой, описывается выражением p (V 2 – V 1) + We, где первый член – работа расширения, связанная с изменением объема от V 1 до V 2, а We –дополнительная, или т.н. «полезная», работа, совершенная системой помимо работы расширения. Если работа совершается над системой, то оба члена имеют отрицательный знак. Поэтому уравнение (1) можно преобразовать к виду
Вводят вспомогательную меру энергии системы Н, определяемую общим соотношением
Если давление постоянно (обычно в качестве стандартного берется давление 1 атм), то изменение функции Н, называемой энтальпией системы, отличается от изменения ее внутренней энергии на величину работы расширения:
За исключением газофазных систем, это отличие пренебрежимо мало по сравнению с типичными тепловыми эффектами реакций. Однако для общего случая из формулы (2) следует, что теплота Q, измеренная при постоянном давлении и We = 0 (обычное условие протекания химической реакции, если она не происходит, например, в аккумуляторе или гальваническом элементе), равна изменению энтальпии системы:
В любом случае, поскольку разность H 2 - H 1, как и U 2 - U 1 , определяется, согласно первому началу термодинамики, исключительно начальным и конечным состояниями системы и не зависит от способа перехода из начального состояния в конечное, суммарное количество теплоты, поглощенное в процессе химического превращения при постоянных температуре и давлении (при We = 0), зависит только от исходных реагентов и конечных продуктов и не зависит от того, через какие промежуточные стадии протекает реакция. Этот вывод был сделан Г.И.Гессом в 1840 на основе экспериментальных фактов еще до классических опытов Джоуля, продемонстрировавших эквивалентность теплоты и других форм энергии. Гесс показал, что теплота реакции, протекающей через несколько последовательных стадий, равна алгебраической сумме теплот отдельных промежуточных реакций. Закон Гесса, как отметил Г.Гельмгольц в 1847, служит прямым экспериментальным подтверждением применимости закона сохранения энергии к энергетике химических реакций. В пределах ограничений, налагаемых уравнением (5), этот закон был многократно подтвержден многочисленными дальнейшими исследованиями. Термохимические уравнения. Теплота, высвобождаемая или поглощаемая конкретной химической реакцией, пропорциональна степени превращения реагентов, определяемой по количеству любого из расходуемых либо образующихся продуктов. Изменение внутренней энергии или энтальпии реагирующей системы определяют по химическому уравнению реакции. Например, сгорание смеси газообразных метана и кислорода описывается термохимическим уравнением
Здесь буквы в скобках обозначают агрегатные состояния веществ (газ или жидкость). Символом D H ° обозначается изменение энтальпии в химическом превращении при стандартных давлении 1 атм и температуре 298 K (25° С) (знак градуса в верхнем индексе H указывает, что данная величина относится к веществам в стандартных состояниях (при p = 1 атм и T = 298 K)). Химическая формула каждого вещества в таком уравнении обозначает вполне определенное количество вещества, а именно его молекулярную массу, выраженную в граммах. Молекулярная масса получается сложением атомных масс всех элементов, входящих в формулу, с коэффициентами, равными числу атомов данного элемента в молекуле. Молекулярная масса метана равна 16,042, и, согласно предыдущему уравнению, при сгорании 16,042 г (1 моля) метана получаются продукты, энтальпия которых на 212,798 ккал меньше энтальпии реагентов. В соответствии с уравнением (5) такое количество теплоты высвобождается, когда 1 моль метана сгорает в кислороде при постоянном давлении 1 атм. Соответствующее уменьшение внутренней энергии системы в ходе реакции составляет 211,615 ккал. Разница между D H ° и D U ° равна -1,183 ккал и представляет работу p (V 2 - V 1), совершаемую, когда 3 моля газообразных реагентов сжимаются при давлении 1 атм до 1 моля газообразного диоксида углерода и 2 молей жидкой воды. Стандартная теплота образования. Из закона сохранения энергии следует, что, когда вещество образуется из атомов и (или) более простых веществ, внутренняя энергия или энтальпия системы меняется на определенную величину, называемую теплотой образования данного вещества. Теплота образования может быть определена различными способами, в том числе прямыми калориметрическими измерениями и путем косвенного расчета (на основе закона Гесса) из теплоты реакции, в которой участвует данное вещество. При проведении расчетов пользуются стандартными (при p = 1 атм и T = 298 K) теплотами образования веществ, входящих в уравнение реакции. Например, стандартную теплоту (энтальпию) образования метана можно вычислить с помощью термохимического уравнения
Хотя эта реакция практически неосуществима при 25° С, стандартная теплота образования метана косвенно рассчитывается по измеренным теплотам сгорания метана, водорода и графита. На основе закона Гесса устанавливается, что теплота реакции равна разности между теплотами сгорания веществ, указанных в левой части уравнения, и теплотами сгорания веществ, указанных в правой части уравнения реакции (взятых с соответствующими знаками и стехиометрическими коэффициентами). Помимо использования термохимических данных для решения проблем практического использования тепловой энергии, они широко применяются при теоретической оценке энергий химических связей. Этот вопрос подробно рассмотрен Л.Полингом в книге Природа химической связи (The Nature of the Chemical Bond, 1960). Второе начало термодинамики. Второе начало термодинамики по существу определяет однонаправленность переноса теплоты в разнообразных процессах, происходящих спонтанно при определенных условиях, а именно – направление переноса теплоты от тел с высокой температурой к телам с низкой температурой. Второе начало термодинамики может быть сформулировано следующим образом: не может быть спонтанного общего переноса теплоты от тел менее нагретых к телам более нагретым. Перенос теплоты Q от источника с температурой Т можно охарактеризовать величиной Q / T. Для любого спонтанного процесса переноса теплоты, при котором источник с температурой Т 1 отдает количество теплоты Q 1, а в результате переноса система с температурой Т 2 получает количество теплоты Q 2, справедливо неравенство Клаузиуса Q 1/ Т 1£ Q 2 / Т 2. Таким образом, для того чтобы мог происходить перенос теплоты, Т 1 должна быть больше Т 2. Для перехода системы из одного состояния в другое более общая формулировка второго начала термодинамики гласит, что направление переноса теплоты определяется условием
где S 2 - S 1 – разность энтропий системы в двух состояниях. Если скомбинировать это условие с уравнениями (2) и (3), получим соотношение, имеющее важное значение для описания химической реакции при постоянных температуре и давлении:
Если ввести функцию состояния системы
то формулировка второго начала термодинамики примет следующую форму:
Это значит, что для системы, находящейся при постоянных температуре и давлении, могут происходить только такие переходы из одного состояния в другое, для которых полезная работа We не превышает определенной максимальной величины, равной разности D G двух значений G. Если G 1 > G 2, то переход из состояния 1 в состояние 2 (скажем, от реагентов к продуктам) может происходить самопроизвольно даже при We = 0. Если G 2 > G 1, то переход из состояния 1 в состояние 2 может быть осуществлен только за счет внешней полезной работы; это значит, что работа We должна быть отрицательной величиной, как, например, электрическая энергия, затрачиваемая при электролитическом разложении воды. Если G 1 = G 2, то система находится в равновесии. Функция G называется гиббсовой энергией, или изобарно-изотермическим потенциалом. Разнообразными методами было показано, что величина D G °, «стандартная гиббсова энергия образования», по аналогии со стандартной энтальпией образования может быть определена для химических соединений относительно элементов из данных по химическим равновесиям и химическим процессам. Стандартная гиббсова энергия образования D G °, характеризующая какую-либо химическую реакцию, может быть установлена с помощью таблиц стандартных гиббсовых энергий образования путем вычитания суммы их значений для реагентов из суммы значений для продуктов. Значения D G ° для чистых химических элементов при 25° С и давлении 1 атм принимаются равными нулю. Стандартная гиббсова энергия химической реакции по существу является мерой того, насколько реагенты и продукты далеки от равновесия друг с другом при данной температуре и стандартном давлении 1 атм. Согласно второму началу термодинамики, все самопроизвольные изменения системы и ее окружения стремятся привести их в конечное состояние равновесия. Следовательно, именно изменение гиббсовой энергии, а не изменение энтальпии или внутренней энергии определяет возможность протекания химической реакции. В частности, от изменения гиббсовой энергии в ходе химической реакции зависит разность потенциалов между электродами химических источников тока. Стандартное изменение гиббсовой энергии связано со стандартным изменением энтальпии, согласно (7), соотношением
Можно показать, что функциональная связь между D G ° для данной химической реакции и константой равновесия К, определяемой законом действующих масс, имеет вид
где R – универсальная газовая постоянная, равная 1,987×10-3 ккал/(K ×моль). С учетом формулы (9) это соотношение можно представить в эквивалентной форме:
Таким образом, константа равновесия состоит из двух множителей, один из которых содержит энтальпию реакции (или, при определенных условиях, внутреннюю энергию реакции), а другой – энтропию. Важное следствие общей термодинамической теории состоит в том, что можно вывести уравнение, связывающее зависимость D G ° (и следовательно, К) от температуры со значением D H °. Поэтому можно точно установить направление и равновесное состояние химической реакции при одной температуре из известных данных по химическому равновесию при другой температуре. В частности, изменение D G ° с температурой пропорционально величине D H ° и имеет противоположный знак. Так, все эндотермические реакции (D H ° положительна) ускоряются (большая отрицательная величина D G ° и большее значение К) с ростом температуры. Соответственно все экзотермические реакции замедляются с ростом температуры. Третье начало термодинамики. Еще один важный принцип химической термодинамики был установлен в первой четверти 20 в. В. Нернстом. Он экспериментально установил, что, когда температура приближается к абсолютному нулю, стандартные энтропии D S ° для многих химических реакций стремятся к нулю. Поскольку этот результат не следует из первого и второго начал термодинамики, он получил название третьего начала термодинамики. Оно не имеет столь же общего характера, как первые два начала, однако большинство очевидных исключений или аномалий получило удовлетворительное объяснение с учетом особенностей кристаллического строения конкретных веществ. Значение принципа Нернста состоит в том, что зависимость энтропии реакции от температуры может быть выведена, согласно термодинамической теории, исключительно из данных по теплоемкости для отдельных участвующих в реакции веществ. Это значит, что, если известно значение D S ° при какой-либо одной температуре (при T = 0 D S ° = 0), значения энтропии реакции при других температурах вычисляются исключительно из тепловых измерений. Аналогично этому, величина D H °может быть получена из калориметрических данных путем непосредственного измерения теплоты данной реакции либо косвенно, путем измерения теплот других реакций и применения закона Гесса. Следовательно, подстановка полученных значений D H ° и D S ° в уравнение (9) или (11) дает гиббсову энергию или константу химического равновесия исключительно из тепловых измерений. Это позволяет предвидеть направление и равновесное состояние химической реакции еще до того, как найден способ ее проведения. Для практического применения третьего начала термодинамики требуются точные результаты измерений теплоемкости вплоть до самых низких доступных температур, желательно до температуры жидкого гелия (около 4 К). Это направление термодинамических исследований химических явлений стимулировали криогенные исследования, выполненные в последние десятилетия В БСЭ сказано, что термодинамические системы определяются, как “ совокупности физических тел, которые могут взаимодействовать энергетически между собой и с другими телами, а также обмениваться с ними веществом “. В этом определении говорится об энергообмене, а теплообмен - это лишь частный случай энергообмена. И поэтому приведенное определение относится к энергодинамическим системам, как к более общему понятию. В современной физике существует следующая классификация термодинамических систем по признаку возможности энергообмена и обмена веществом с окружающей средой или с другими системами: а) Система открытая, если возможен энергообмен и обмен веществом. Закрытые системы дополнительно подразделяются по признаку возможности осуществления энергообмена следующим образом: а) Система замкнутая, если энергообмен возможен, но обмен с внешней средой путем совершения механической работы невозможен. В адиабатной системе рассматривается как обратимый, так и необратимый адиабатный процесс. Обратимый адиабатный процесс называется также изоэнтропийным процессом, чтобы подчеркнуть постоянство энтропии в адиабатной системе. А постоянство энтропии означает отсутствие необратимых диссипативных потерь энергии.
|